Examples
Correlating Optical and Structural Properties of CO on Transition Metal Surfaces
Here we report on an optical study of CO adsorbed on Pt (111) and Cu (111) surfaces, based on the Quasiparticle Self-Consistent GW (QSGW) approximation. It combines structural information taken from density-functional theory (DFT) to elucidate molecular spectral features at this interface.
By varying CO coverage θ, as well as its placement relative to the TM substrate, we can resolve spectral features of both occupied and unoccupied molecular states. Where experimental data is available we show QSGW compares to it very well. Assuming QSGW has comparable fidelity for similar interfaces, its predictive power provides a new path to infer information about the structure of CO from optical information. Further, the theory can help to predict the presence of other little-understood adsorbates such as an OCCO dimer, that may be relevant to mechanistic pathways for reduction of CO2 to high value C2+ products. This new approach complements total energy calculations, and also fills a void in DFT based theory that is known to be an unreliable predictor of the energetics of CO on transition metal surfaces.
CO is an important probe molecule used to study transition metal (TM) active sites for heterogeneous catalysis by infrared spectroscopy. Moreover, CO is a key reactant-product in CO2 reduction to low-carbon fuels and high value products such as alcohols, acids, hydrocarbons and carbon sequestration. It is also an essential component in the initial step for reaction processes such as CO2 splitting into CO* and O*.
Knowing how CO initially binds to the substrate is an essential component for all of these processes. However, our theoretical understanding of CO adsorbed on a TM surface is fragmented. This is largely because our main tool to model such systems— density functional theory (DFT) even with functionals tuned for catalysis—lacks sufficient fidelity to reliably describe either the total energy or spectral properties. As one example, many functionals strongly over-bind CO at the hollow site on TM surfaces.
If already the site preference of the initial step of the reaction process cannot be resolved to the required accuracy, modeling the entire reaction process remains in doubt. The study reported here circumvents this difficulty, and least in part, but focusing on optical properties: optical studies yield different kinds of information about the chemistry of the interface than given by thermodynamics. By correlating DFT-derived structural information with high fidelity calculations of optics, we provide an alternate and surer path to better understand these interfaces. This is possible because, even while DFT energies are not found to be reliable, DFT predictions of the structure are much less uncertain. Detailed studies of structure show that they are not strongly sensitive to choice of density-functional, and also align well with measured values (see the paper’s supporting information).
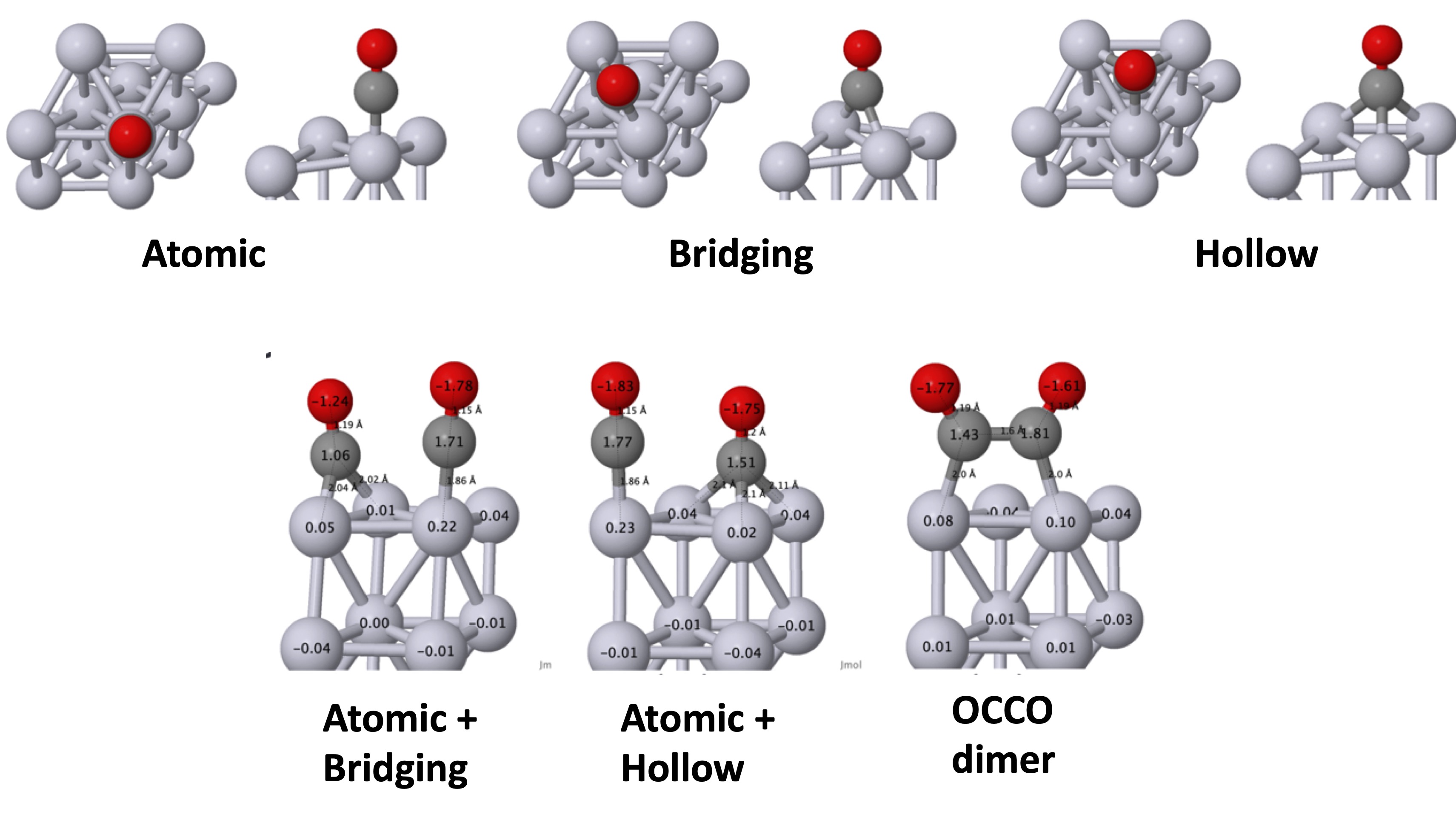
Where is CO placed above the substrate? is the first question that must addressed. There are three high-symmetry positions: atomic, bridging, hollow sites (Fig. 1). On the theoretical side, thermodynamics derived from total energy calculations would yield the relative occupation of different sites if the free energies of the respective sites were reliably known. They are not, however, and it is necessary to rely on experimental information. In-situ scanning tunneling microscopy (STM) is a very useful probe that can identify the ratio and position of atomic, bridging, and hollow sites, and also the importance of CO-CO lateral interactions for higher coverages θ. IR-absorption spectroscopy (IRAS) can yield similar information, but results fromo the two measurements can vary considerably. Since STM is likely the more reliable measure, some caution is warranted in inferring coverage from IR data.
Site occupation probabilities (and the CO-CO lateral interactions) depend on coverage θ as well. The paper considers atomic, bridging, and fcc hollow CO sites at θ = 1/4 coverage, and combinations of them which correspond to experiments at θ = 1/2 coverage. This work elucidates QSGW’s ability to match photoemission data on Pt and Cu substrates with high fidelity, and make it possible to resolve the unique features related to different CO site occupations.
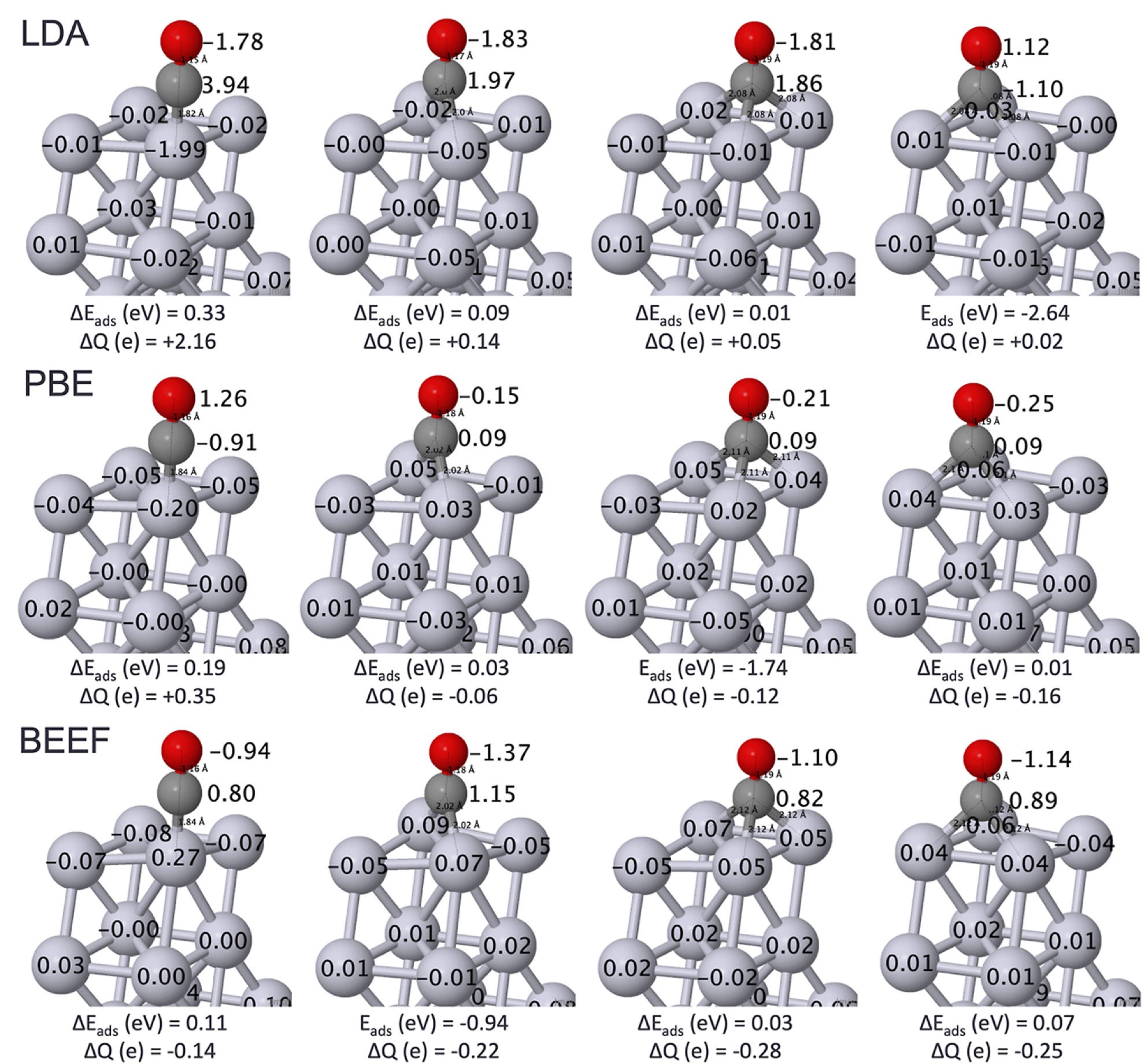
Fig. 2 shows cartoons of the four high-symmetry sites that may occur at low converage, with the DFT adsorption energies and Baader charges, for three different functionals. Baader charges vary wildly across functionals, and since the energy involves integrations of density-related objects, it is a warning that energy functionals are also unreliable. Indeed variations total energies are not small on the relevant energy scale (kBT). On the other hand, the different functionals predict similar structures, and they moreover correspond well to experimental data. So taking structural information from DFT is justified.
Partial DOS from the Quasiparticle Self-Consistent GW approximation.
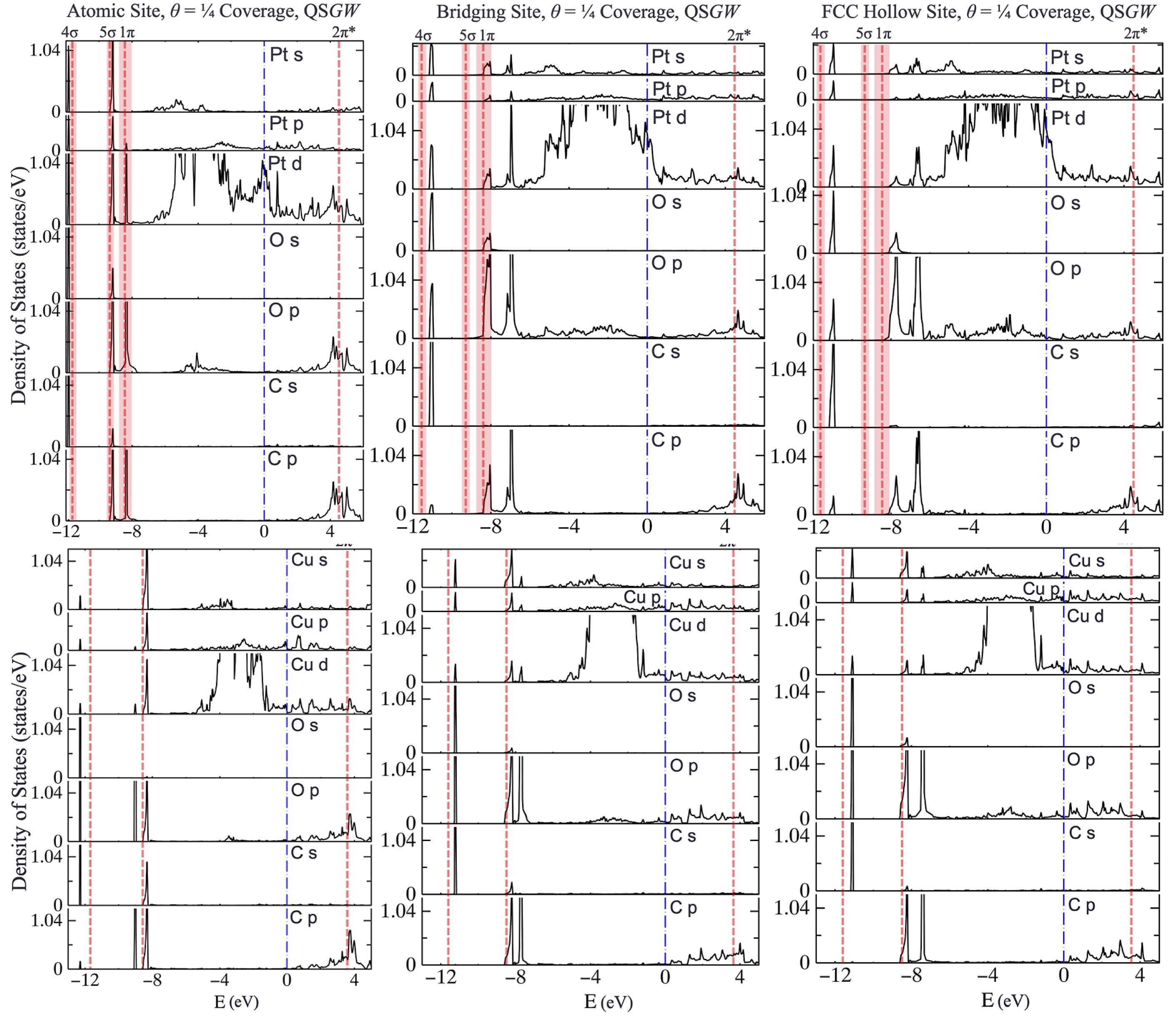
Top panels of Fig. 3 show the QSGW partial DOS for CO on Pt(111) at θ = 1/4 coverage. DOS is resolved by species and orbital Also shown are experimental data from ultraviolet photoemission spectroscopy for occupied states and from a two-photon experiment for the unoccupied 2π* state at around 4.5 eV, for θ=1/2 coverage, as explained in the paper. The experiment and theory are not fully compatible as one is calculated for θ=1/2 and the other at θ=1/4. Nevertheless useful information can be drawn. First the 2π* state, which does not strongly on structure, is very well desribed by QSGW. However, the three bonding states (4σ, 5σ, 1π), do depend in a significant way. Which of the three is more likely depends on the occupation probabilities. There is some uncertainty in the relative energies of the three sites, but the ranges of values suggest that the atomic site is 5-100 times more likely to be occupied than the others. If the atomic site is dominant, QSGW DOS align very well with PE data for all three levels 4σ, 4σ, 1π (left panel of Fig. 3). The DFT DOS, do not correspond well to experiment for any geometry (see paper).
Less experimental information is available for CO on Cu(111) (bottom panels of Fig. 3), but useful comparisons can be made. In many respects the Pt and Cu cases are very similar, but there are some differences. The first is that the Pt d states cut through EF, while the corresponding Cu d states lie mostly below. Regarding the CO levels, two peaks are recorded in the valence for Cu instead of the 3 for the Pt case (4σ, 5σ, 1π). The QSGW noninteracting DOS shows three for Cu, with the 5σ, 1π closely spaced. The proper interacting DOS will broaden relative to the noninteracting one shown, and very probably the 5σ and 1π will merge into one peak, thus explaining why only two levels can be resolved. (The experiment cannot distinguish 5σ and 1π peaks.) Generating the interacting DOS is possible in Questaal, but was not done in this work. How to make it and the distinction between G and G0 is explained in this tutorial.
Some experimental is available for θ=1/3 and for θ=1/2. From IRAS studies the ratio of population of atomic and bridging sites was estimated to be 13:12 (i.e. nearly equal population) for θ=1/2 coverage. (The study also suggests that the hollow site may play a larger role for Cu.) If atomic and bridging sites have equal populations, an average of the QSGW peaks (which fall slightly above the PE data, or slightly below, depending on geometry) the two would align very well with the PE data. The LUMO provides other contrasts between Pt and Cu cases: the 2π* LUMO has a moderately sharp peak only for the atomic site (at 3.6 eV, close to experiment for lower coverages). Additionally, in the Cu case, QSGW predicts the LUMO shifts from 3.6 eV at θ=1/4, to 3.35 eV at θ=1/2. Two-photon photoemission data for θ=1/3 and for θ=1/2 also show a shift from 3.6 eV to 3.35 eV, providing another very useful benchmark for the theory.
This shift, which is a conseqence of CO-CO interactions, appear to be stronger for the CO adsorbed on Cu than on Pt. This may be important for chemical reactions, as CO-CO interactions (possibly involving the OCCO dimer, though this is unresolved) could play a role in the selectivity of reaction products.
PAPERS · CATALYSIS · TRANSITION METAL SURFACES · OPTICAL PROPERTIES